What Are Clinical Trials?
Clinical trials are a type of clinical research that where researchers study specific illnesses and the safety and effectiveness of new interventions (such as drugs, treatments, diagnostic mechanisms, or devices). Before an organization can release one of these interventions into the consumer marketplace or use it in clinical practice, researchers must conduct extensive research, including clinical trials. This research is mandatory and scientists conduct it in steps and stages.
In the United States, the Food and Drug Administration (FDA) evaluates the research and outcomes of clinical trials via their principles of good clinical practice (GCP). In the European Union, the European Medicines Agency (EMA) sets drug licensing; in the UK, the Medicines and Healthcare Products Regulatory Agency (MHRA) licenses drugs. In Canada, Health Canada reviews the efficacy and safety of drugs before they can be sold. These agencies represent different countries and regions, but have highly similar functions.
Organizations develop clinical trials to show what works best for specific people or illnesses. In the U.S., the FDA approves drugs for specific illnesses and conditions, but once a drug is released into the marketplace, medical professionals can legally prescribe for other illnesses or conditions. The FDA regulates the marketing for these drugs, such as the claims made in advertising, but does not regulate the prescribing of drugs by medical professionals. Approved drugs prescribed for conditions or illnesses for which they have not been tested are called off-label prescriptions. Off-label prescriptions are common and, according to one study in 2015, account for 10-20 percent of all prescriptions.
Researchers evaluate many other medical issues in clinical trials, including the following: medication combinations, new uses for existing medications, new types of surgery, new medical equipment, and treatment protocols. Investigators study all of these issues in order to gain information on efficacy, toxicity, and pharmacokinetics. In other words, researchers want to know if drugs or treatments work as intended, if and at what level a medication becomes toxic, and how a drug reacts specifically in the body.
In some cancer clinical trials, the goal is remission, which means that there are no more cancer cells detectable in the body. The research process leading up to clinical trials often includes animals (in vivo studies) and extensive laboratory work (in vitro studies). A drug is usually prescribed to treat a specific illness or condition.
Funding Clinical Trials
Clinical research funding comes from several sources, such as federal institutions, like the National Institutes of Health (NIH), the Department of Defense (DOD), and the Department of Veterans Affairs. Private industry, such as biotechnology and pharmaceutical companies, as well as medical centers, foundations, and universities, also fund studies. This funding covers the basic research of a clinical trial, such as the salaries of researchers and any materials, therapeutics, and equipment involved.
Pharmaceutical companies fund most clinical trials for new drugs, even if the study is in cooperation with or spearheaded by a medical institution or university. In 2007 (in the most recent study of its kind), researchers reported that almost 75 percent of funding for clinical trials in the U.S. came from corporate sponsors.
The Development Process of Clinical Trials
The attempt to discover a viable new medication, treatment, or therapy is known as the discovery process. Scientists working in research and development can begin this journey in any number of ways. New insights into a disease process can allow researchers to design a new product that eradicates the effects of that disease. Tests can confirm that certain molecular compounds have beneficial results against certain diseases. Pre-existing treatments can have unintended consequences. Scientists can develop novel technology, allowing medical interventions to target specific sites in the body or genetic material.
After the discovery process begins, researchers move on to development. If they identify a promising intervention during this stage, they move on to experimentation. During experimentation, scientists gather the following kinds of information:
- Mechanisms of action
- Possible benefits
- How the body absorbs, metabolizes, and excretes a drug
- Potential routes of administration
- How a drug interacts with other medications
- How one can produce a drug for administration
After development, researchers perform preclinical research, using good laboratory practices (GLP). Scientists do not perform these studies on humans, but the results should yield useful information about dosing and toxicity levels. Following the preclinical work, investigators perform the clinical research. The FDA monitors these clinical processes. They also oversee the succeeding steps in a drug’s progress, including the drug review and the post-market drug safety review for a minimum of two years (and, in some cases, longer) after a new treatment is released. The FDA may remove a treatment from the market anytime it discovers evidence that the treatment is a human safety risk. The FDA puts out regular updates and recalls treatments regularly. The agency is also developing a real-time surveillance system called Sentinel that supports adverse event reporting.
Types of Clinical Trials
Clinical trials come in two main varieties:
- Observational: This type of clinical trial monitors conditions and health over time. The patients in these trials may be receiving treatment, but they are not assigned to specific interventions. The data collected from these studies advances researchers’ understanding of a condition and its treatments.
- Interventional: These are what most people think of when they think of clinical trials — the studies that assign interventions, such as drugs or experimental treatments, to patients with an illness or condition.
There are two types of interventional studies: controlled (or comparative) studies and open-label studies. Controlled studies assign one group the intervention and the other group a placebo. A placebo patient receives the same protocol as the other patients, whether that be a pill, injection, or infusion, but without the active ingredient that the trial is studying. Often, studies that are controlled are randomized as well. Randomized controlled trials (RCTs) randomly allocate patients in trials. Often, the researchers use a computer program to randomize participants into distinct groups. This kind of randomization ensures that the research team’s opinions do not affect who gets which treatment. This method, thereby, decreases bias as much as possible. RCTs are often considered the gold standard of medical intervention studies.
Open-label studies, on the other hand, are transparent (or “open”) regarding which patients are receiving the active drug and which are receiving a placebo. Critics of this type of trial argue that excessive bias is built in to the study’s design.
Often, researchers and politicians distinguish between efficacy trials and effectiveness trials. Efficacy trials are explanatory trials meant to discover whether an intervention yields the expected results under ideal circumstances. Effectiveness trials are pragmatic trials that measure an intervention’s degree of positive effect in real-world circumstances. The study’s design and hypotheses are based on routine clinical practice conditions. Scientists can also assign clinical trials to the following study categories:
- Treatment Trials: These trials test medicine or treatment protocols on patients with a specific illness.
- Prevention Trials: These trials test such preventive measures as vaccines or interventions that may lower the risk for developing a specific condition or illness. Prevention trials may be broken down into action or agent studies. In action trials, the participant “does” something to prevent a disease; in agent trials, the participant “takes” something to prevent a disease.
- Screening Trials: These trials test ways of detecting an illness or condition, especially in its preliminary stages.
- Palliative Trials: These trials look for protocols or approaches to improve the comfort or quality of life of someone living with an illness or condition.
- Genetic Trials: These trials look for the inherited risk of a condition or illness.
The literature on clinical research describes several types of studies that have published results:
- Case Series: Also called clinical series, these observational studies track patients who have the designated condition or illness. Often considered half of a case-control study, case series alone have no control group. (The members of a control group are not afflicted with the condition or illness under study.) The patients in case series studies often receive the same intervention. Case series studies usually describe these patients, what treatments have been performed, and what their outcomes look like without much comparison between them or conclusions being drawn in the study report.
- Cohort Studies: In contrast with case series, cohort studies have a defined group of people (a cohort) who are followed over time (for their designated illness or condition and their treatment) and compared to each other. For example, a cohort study design could look at two groups of patients, both with the same illness but receiving two different treatments to see if one works better than the other. Researchers say that it is often difficult to determine the difference between case series and cohort studies because they are often designed so similarly, and some publication authors do not categorize their studies as either type.
Another way to designate a type of clinical trial is whether it is a multicenter or single-center trial. Multicenter or multi-site trials are the same trial offered at more than one clinic or medical center. Large trials, particularly those in Phase III, offer more flexibility for patients and ability for the study to garner more participants and variability when it is offered in more than one location. Further, some studies compare several treatments at once. Called multi-arm multi-stage (MAMS) trials, there may be three or more treatment groups testing different interventions, and they may shut one treatment arm down if the early results do not show a treatment is working or has more side effects.
Medical device clinical trials are unique because they are smaller in scale overall and require less phases. Medical device studies are more difficult to blind, randomize, and control. Many are dependent on physician’s technique, and device modifications usually happen during the trial. It may be impossible or unethical to use a placebo in a medical device study.
Not all medical devices require a clinical trial for release. There are three different classes of medical devices: I, II, and III. The class types escalate with the risk to the patient of the device.
- Class I has low to moderate risk, comprise 47 percent of devices, and include things such as bandages, gloves, bedpans, and manual stethoscopes.
- Class II are moderate to elevated risk for the patient, comprise 43 percent of devices, and can include things such as single-use scalpels or pregnancy test kits.
- Class III medical devices are the highest risk to the patient and are usually implanted or support life. Examples of these are breast implants or pacemakers.
There are two type of medical devices clinical trials:
- Early Feasibility or Feasibility Study (EFS): This small study is designed to get insights into a novel medical technology during development before the larger clinical trial. These studies are optional but involve the FDA early to streamline clinical testing later. In a traditional feasibility study, formal endpoints (also called possible outcomes) are determined, and would inform future testing.
- Pivotal Study: This is an FDA term, and is not exclusive to medical device studies. For medical devices, however, the process is sped up considerably, so the pivotal study comes much earlier than it would be required for a drug or vaccine study. The pivotal study is the large, human study that directly precedes a marketing application.
Designing Clinical Trials
Researchers design a clinical trial to follow a specific study plan or protocol. This helps them answer specific research questions about their studies’ intervention. A well-designed study plan helps secure funding, ethics approvals, regulatory approvals, and any needed development approvals. Professionals recommend getting statisticians involved in the planning process to help you with design: choosing an appropriate outcome, sample size, randomization methods, analysis plan, data collection tools, and interim data reports. Good protocol plans have the following:
- A simple method targeted to the patient group, delineating the selection criteria, number of participants needed, study length, and drug administration and dosage (where applicable)
- A research question and objectives that are clinically relevant and not addressed already
- Justified data requirements
- The most reasonable choice of controls (where applicable)
- Allocation concealment (where applicable)
- Blinding procedures for the intervention and the outcome assessments (where applicable)
- An explanation of how research bias will be confined
For drug trials, the sponsors (or the development company) must apply for an investigational new drug (IND). This application includes the following: the data detailing the effects of the animal trials, the manufacturing information, the study plan, prior human research data, and the principle investigator credentials. The FDA helps for free when drug companies ask for help during any part of the drug development process. This can include before the IND is submitted and during some of the clinical trials phases.
The FDA has a review team for each IND application they receive, consisting of scientific specialists who are responsible for distinct parts of the review. This process is meant to protect clinical trial participants from excessive risks. These teams include the following:
- Project Manager: This is the FDA’s main internal application coordinator and the main point for contact for the applicant (or sponsor).
- Medical Officer: This team member reviews all the study’s clinical data during all study intervals.
- Statistician: The statistician on the team is responsible for reviewing the study design and data and working with the medical officer to ensure the protocol is safe and efficacious.
- Pharmacologist: This member reviews any preclinical study data.
- Pharmacokineticist: The pharmacokineticist is responsible for regular review of the data on the drug’s effect in the body, including whether the dosages and administration schedules are appropriate.
- Chemist: This is the person responsible for understanding the drug’s chemical composition, how it was made, its stability, whether there is good quality control during manufacture, and whether there are any impurities present.
- Microbiologist: If the drug is an antimicrobial, this person reviews the data submitted to see the microbial response.
For IND submissions, the FDA has 30 days to respond. They either approve the application or put a hold on or completely stop the investigation. Although rare, a clinical hold could be because of exceptional risk, unqualified investigators, misleading participant marketing, or lack of information about risks. In these cases, the FDA will respond with comments about what they need to continue their review. The investigators are responsible for communicating any protocol changes, significant side effects, and reporting to the review team. This process continues through the trial and until its end unless a marketing application is filed. Two large controlled trials worth of data must be submitted with a marketing application. For medical devices, researchers would request an investigational device exemption (IDE) for their application, as an IND is not obligatory. An IDE is used so that clinical data can be gathered on a medical device without the same procedural requirements that drugs, vaccines, or cancer protocols must go through.
What Is a Clinical Trial Phase?
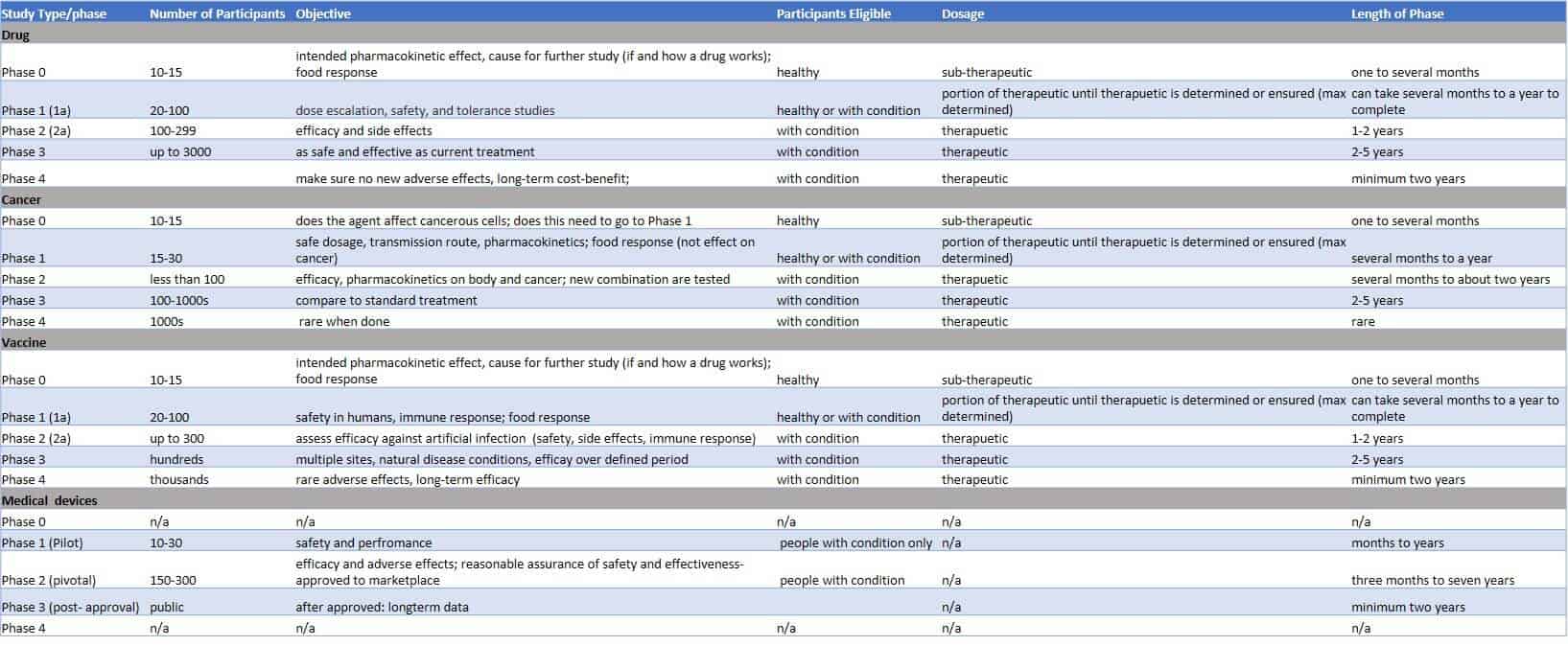
A clinical trial phase is one of the stages of testing that a new intervention, whether a drug, treatment, or device, goes through to become available for legal use. These phases normally consist of phase I through phase IV, but can also include a phase 0. The phase describes the purpose of the clinical trial and how many participants are involved.
Phases are different from cancer stages and do not match the stages of cancer. Cancer patients can have any stage of cancer and be in any phase of clinical trial.
Once the FDA approves, whether via an IND or an IDE, intervention testing on humans can begin. Four clinical trial phases are typical, with each being considered a separate trial with a different purpose. After each phase is completed, the FDA’s review team must evaluate the data before the study can move onto the next phase. The phases have some slight variations based on the type of clinical trial being conducted (i.e., whether for drug, vaccine, medical device, or cancer protocol).
Sometimes, phases in cancer clinical trials are combined. For example, phases I and II are combined with the aim of finding the highest safe dosage (phase I) and to see how well that dosage works (phase II). This combination can improve the transition time between phases when researchers are trying to expedite the process. This would be written as “phase I/II” in a trial.
An IDE allows one pivotal trial following the feasibility stage: The study must be a reasonable assurance of safety and effectiveness for when the sponsors submit the marketing application.
According to researchers, a new drug typically takes anywhere from 10-18 years to develop and costs between $800 million and over $1 billion. In 2014, the Tufts Center for the Study of Drug Development reported that the cost for them to develop a new drug is about $2.6 billion, due to the high failure rate of seven out of eight attempts. Currently, in the U.S., the FDA approves only about 10 percent of new drugs applied for with an IND. The best rates of success are often for drugs that target blood-related conditions, according to a report of clinical development success rates: 26.1 percent of medications developed for hemophilia, anemia, and blood protein deficiencies are eventually approved. About five percent of oncology drugs are approved, and about 9.6 percent of neurology drugs are approved.
What Is Clinical Trial Phase 0?
Clinical trial phase 0 comprises any and all preclinical studies. Preclinical studies must exist prior to the beginning of or even application for any clinical trial. These studies use preliminary data from laboratory work and animal studies. Some of these studies (called in silico studies) even use computer modeling for the drug-target interactions. Toxicology studies are also performed preclinically.
The point of preclinical work is to determine whether an intervention will move to the clinical trial stage. Critics say that the preclinical stage is often misleading as to whether new agents will work, because the stage can yield both false positive and false negative results.
The FDA introduced phase 0 in clinical trials in January 2006. Phase 0 clinical trials, sometimes called proof-of-concept trials or pre-phase-I trials, are not like other phases of clinical trials. Phase 0 in a clinical trial is an exploratory, first-in-human (FIH) trial run, according to the FDA’s guidelines. To obtain approval from the FDA to begin phase 0 studies, you require less preclinical data than you do for phase I studies. Researchers can run phase 0 studies simultaneously with the generation of preclinical toxicology data. These toxicology studies are often called micro-dose studies, because they call for giving only one sub-therapeutic dose to each member of a very small group (10-15) of otherwise healthy people.
Often, companies run phase 0 studies because they are trying to decide between candidates for further research and development. Another typical reason to run a phase 0 study is to expedite the drug approval process for new molecular medications. This phase usually yields the following outcomes: the generation of data about how the drug works in the body and the proof that the product demonstrates the intended drug-target effects. In cancer studies, phase 0 helps keep negative side effects at bay for many study participants, while also determining whether the agent affects cancerous tumors and merits further study.
Phase 0 studies can save organizations time and money by allowing researchers to weed out unrealistic candidates much earlier in the process via the failure of new agents. These preliminary studies can also shorten the phase I study by discovering early on the appropriate therapeutic dosage for novel medications. Phase 0 studies are not common in cancer trials and are rare in vaccine or medical device trials. Phase 0 is not a required part of testing a new intervention.
What Is Clinical Trial Phase I?
Phase I clinical trials are usually the first real stage of testing on humans. The goal of this study phase is to determine the highest dose of a new treatment that may be administered without serious side effects. Even though at this point, animal testing and laboratory assays have already been completed, testing on humans is not always predictable, as their physiology may react differently and produce unforeseen side effects.
Researchers also use phase I studies to determine the best route of administration. Placebos are not used in phase I studies. Phase I usually tests about 20-100 people with the new intervention; these study participants can either be healthy or afflicted with the illness or condition under consideration. Sometimes, those with said condition can participate because they have tried and failed to improve after being treated with traditional therapies. (This kind of study occurs especially with cases of HIV and cancer.) The people who volunteer in this study phase are usually paid.
Phase I is the stage during which you slowly increase a drug’s dosage in order to study the side effects. You treat the volunteers in a clinic, so you can closely observe them and provide medical attention until a drug’s half-life has passed. A drug half-life is a term in biological science that refers to the exponential rate of removal in the body of a substance. Put simply, the term refers to the body’s ability to repeatedly rid itself of half of a drug. For example, if the dosage is 100 mg, then its half-life is the amount of time it takes until only 50 mg are still present in the blood or plasma. If the half-life is two hours, then the research team calculates how many two-hour intervals it takes until only a clinically insignificant dosage remains in the body. The researchers monitor the participant until this happens.
The goal of phase I is called dose escalation, i.e., getting to the best dose with the fewest side effects. Dose escalation also allows you to determine how often and when people can take a drug. In phase I, one calculates dose escalation based on the dosage that was toxic in the animal studies. During this phase, one also studies the effects of combining food with intervention administration. The study reviews what effect — if any — food has on the body’s absorption of the medication. Groups that eat before the administration of a drug are compared to those that do not eat before the administration of a drug. In phase I vaccine studies, the researchers also study the effect on the immune system. In medical device studies, scientists refer to phase I as the pilot phase. This is the time during which you evaluate the device for safety and performance. In medical device studies, only people with the condition under consideration can be subjects.
You can further divide phase I into phase Ia and phase Ib. The typical phase I study consists of multiple separate trials.
Phase Ia is a single ascending dose study. In this type of study, you give the participant a single dose of an intervention and closely observe them. When there are no adverse side effects and you get confirmation of the expected pharmacokinetic activity, you escalate the dose. Then, you test new participants with this new, escalated dose. You repeat this process iteratively until you reach the maximum tolerated dose (MTD). In the case of adverse side effects, you decrease the dosage and then determine the MTD. You have officially reached the MTD when one third of participants exhibit severe side effects.
Phase Ib is a multiple ascending dose study. Considered a faster route than the previous phase I studies, Ib tests various drug dosages. It accomplishes this by simultaneously administering different drug dosages to each branch of the study, while maintaining one protocol throughout. For example, one third of the participants would receive the new agent at one third of what is assumed to be the clinically significant research dose; one third would receive the new agent at two thirds of the predetermined dose, and the remaining one third would receive the full dose presumed to be clinically significant. Scientists also study the blood or plasma samples of the participants in order to yield information about how the body is processing the drug.
Phase I studies, whether Ia or Ib, are the studies that pose the most risk to the participants. However, these studies can also alleviate a person’s condition or illness, so prospective participants should closely consider the potential risk-benefit ratio. Phase I studies generally take several months to a year to complete. About 70 percent of new drugs move from phase I to phase II.
What Is Clinical Trial Phase II?
Once phase I ensures a drug’s safety by setting the clinical dosage and the MTD dosage, testers can initiate phase II. During this phase, you continue to study drug safety. However, phase II is primarily concerned with the next big goal for bringing a new intervention to the marketplace: seeing if a drug actually works as intended.
The goal of the testing is dependent on the goal of the treatment. Researchers try to figure out if a drug works at all or if it has a minimum level of biological activity. For example, if a drug has an activity level at an estimated 10-20 percent of the MTD, the sponsors may not feel that it’s worth further funding, as the adverse side effects might outweigh the clinical utility. In this case, the drug would not move to phase III.
If there is suspicion regarding metabolic rate variations, genetic testing may take place during phase II. Some phase II studies are designed as case series, and some are designed as RCTs. Case series demonstrate the safety and activity of a drug in the selected patients. RCTs demonstrate a drug’s effectiveness as compared to that in control group (placebo) patients.
In cancer studies (wherein the type of cancer is the same for all participants), scientists often study new combinations of therapies. One does not use placebos in cancer phase II clinical trials.
In vaccine studies, phase II looks at whether an agent is effective against artificial infection and what the participants’ immune response may be.
Phase II medical device studies are called pivotal studies, because they represent the line between an experimental device and its release into clinical use. The purpose of a phase II study for a medical device is to reasonably ensure the device’s safety and effectiveness so when the sponsor submits a marketing application, the application is approved.
Phase II studies usually require populations of approximately 300 participants. (That number pertains to medical device studies as well as most other types of phase II studies.) In phase II cancer studies, however, there are usually less than 100 participants. In phase II studies, only patients afflicted with the illness or condition can participate. This particular restriction makes phase II clinical trials more challenging than earlier phases.
Recently, there has been a push toward the standardization of dividing phase II into phase IIa and phase IIb, especially concerning HIV vaccine and drug trials. Phase IIa includes all those activities normally associated with phase II of a clinical trial. The IIb clinical trial phase is an additional step preceding phase III that targets dose-efficacy — essentially, a small scale efficacy trial that will save money in the long run if the product does not work out.
When the development process for a new drug fails, it is often during phase II trials. Phase II has the lowest success rate of any of the phases: Only 18-33 percent of drugs move on to phase III studies. Phase II studies can cost up to $20 million.
What Is Clinical Trial Phase III?
Generally, less than one third of drug investigations reach phase III. Phase III guarantees that a drug is safe and effective for a specific disease or condition and takes research a step further by delineating that drug’s effect in clinical practice. This study phase is the definitive trial for a drug’s effectiveness, because it compares the drug’s performance with that of the current standard treatment. Researchers can use placebos during this phase when there is no standard treatment.
At a minimum, the treatment must be as safe and effective as what is currently available for that disease or condition. Phase III trials are the longest and most expensive phase and are often RCT multicenter trials, with one branch receiving the standard care and another receiving the new intervention. These RCT studies are often performed with the gold standard of study, the double-blind trial. In a double-blind trial, neither the investigator nor the participant knows which of the treatments the participant is getting.
Phase III usually involves up to 3,000 participants who have a disease or condition, with cancer studies being a bit smaller, depending on the type of cancer. In cancer phase III studies, the trials are usually performed simultaneously across the country by community-based oncologists. In vaccine clinical trials, phase III tests the efficacy of a vaccine against a disease’s natural conditions and reviews how the vaccine holds up over a defined period.
This phase of clinical trials can take from two to five years. It must combine data from two completed trials (into one paper) before the FDA considers moving the product on to phase IV and marketing.
Medical device phase III clinical trials are different than general phase III trials. In medical device clinical trials, phase III is called the post-approval phase, because it occurs post-release of the device’s approval for clinical use. During this phase, investigators collect the long-term data and submit it to everyone who uses the device.
Some manufacturers speed up phase III’s release process by starting the process of gaining regulatory approval. They must submit all manufacturing, preclinical, and clinical data to get this approval, and the drug may be pulled if any information on adverse effects is reported. It is also common for the phase III trial to continue as the regulatory agency sorts out the approval practice to keep patients on the drugs until they can purchase them legally.
Sometimes, the manufacturer will apply for and run label expansion studies, officially called IIIb studies. Label-expansion studies show the regulatory agency that the treatment could work for additional types of patients or diseases beyond the original use. Successful label-expansion studies mean that the company can market the drug for these other diseases or populations. Once phase III is officially complete, the sponsor or pharmaceutical company can request approval to market the drug.
There is some debate about how successful phase III clinical trials are overall. The FDA says that approximately 25-30 percent of drugs move from phase III to phase IV, but some commercial sites say that 70–90 percent move from phase III to phase IV. The average time between filing for approval in phase III to receiving it from the FDA is 1.6 years, with cancer drugs having the shortest approval time (about 1.1 years), and neurological drugs having the longest approval time (about two years). Phase III clinical trials can cost as much as $53 million.
What Is Clinical Trial Phase IV?
Phase IV clinical trials are also known as post-marketing surveillance trials or confirmatory testing. They are conducted after the FDA has approved a drug for commercial sale. Although at this point researchers have already tested a new drug on thousands of people and received approval, they still may not know its full effects and may have further questions.
For example, in a cancer trial, a drug may have been shown to decrease cancer cells and tumors. However, there may be questions about whether this drug decreases the risk of recurrence or whether there are rare side effects that just haven’t surfaced yet. Phase IV trials are meant to address these types of follow-up questions.
The sponsor’s goal in this phase is to compare the treatment (including its cost) with what is already in the marketplace, monitor its long-term effectiveness, and look for any additional adverse effects. Sometimes, phase IV trials end with the FDA taking a drug or treatment off the market or placing restrictions on a product.
In cancer trials, phase IV studies are uncommon. In vaccine clinical trials, phase IV studies look for the rare adverse effects and the long-term efficacy that may emerge, because viruses and bacteria can evolve over the long haul, sometimes making a vaccine less effective.
For new medicines, phase IV trials can require thousands of participants. In these cases, researchers collect data on a new drug and monitor its performance regarding the disease or condition. Although phase IV is the safest of clinical trials, investigators still continue to review the drug for safety issues.
One may conduct phase IV trials for a variety of other reasons as well: Regulatory agencies may require a sponsor to conduct a phase IV trial; a sponsor may initiate a phase IV trial to collect data on different populations; a sponsor may conduct a trial to study a drug’s interactions with other drugs; or companies may want to review how a drug competes in the marketplace. The minimum period for phase IV is two years.
Topics Related to Clinical Trial Phases
- Health Insurance Coverage of Clinical Trials: The Patient Protection and Affordable Care Act (ACA) says that health care plans and insurers cannot keep their clients from joining clinical trials, limit people who join clinical trials, or deny coverage of regular medical costs or increase costs because someone has joined a clinical trial.
- The Targeted Agent and Profiling Utilization Registry (TAPUR) Study: TAPUR is a clinical trial for adults with late-stage cancer whose standard treatment has not worked or has stopped working, or for cancer patients where there is no standard treatment. This is the first clinical trial conducted by the American Society of Clinical Oncology (ASCO).
- ASCO Annual Meetings: The American Society of Clinical Oncology's (ASCO) has an annual meeting where thousands of scientific abstracts are released.
- ASCO Care and Treatment Recommendations for Patients: Easy-to-read summaries give patients information, recommendations with explanations, and lists of things for patients to discuss with their doctors.
- Health Disparities and Cancer: The differences in cancer occurrence, frequency, burden, and death show the disparities in cancer among different races and ethnicities. The Institute of Medicine discovered overwhelming evidence of these disparities, connected to health care and determined they would develop and implement strategies to decrease and eliminate them. For more information on cancer disparities, see Cancer Health Disparities in The United States: Facts & Figures.
- For Patient Advocates: Cancer advocates are people who support cancer causes and policy development. These people work on the national and local level raising awareness, advancing research, improving the quality of care, and addressing legal issues that affect cancer patient care and research.
- Public Policy Advocacy: There are many projects and issues that require advocacy on the national and local level for cancer topics. Some of the issues represented are access to care, clinical trials, drug shortages, federal funding, interoperability and cancer care, quality of care, and value in care. For more information, see ASCO’s advocacy and policy resources.
- Cancer Awareness Dates: There are several important dates and calendar months dedicated to cancer awareness. For a complete list, see cancer awareness dates.
- ECMC clinical trials: The Experimental Cancer Medicine Centre (ECMC) is a network of special cancer research facilities in the U.K. that bring together cross-discipline researchers and professionals. For more information about these facilities, see ECMC.
Improve Clinical Trials with Smartsheet for Healthcare
Empower your people to go above and beyond with a flexible platform designed to match the needs of your team — and adapt as those needs change.
The Smartsheet platform makes it easy to plan, capture, manage, and report on work from anywhere, helping your team be more effective and get more done. Report on key metrics and get real-time visibility into work as it happens with roll-up reports, dashboards, and automated workflows built to keep your team connected and informed.
When teams have clarity into the work getting done, there’s no telling how much more they can accomplish in the same amount of time. Try Smartsheet for free, today.