What Is Corrective and Preventive Action?
Corrective and preventive actions are processes for identifying, documenting, and addressing defects, deficiencies, and nonconformities.

“CAPA is the immune system of your organization,” explains Nathan Conover, President of the consulting and training firm Pathwise. “It’s a system by which you can identify deviations or quality events, investigate them, and implement corrective and/or preventive actions to improve your organization,” he adds.
What Is CAPA?
CAPA is the abbreviation for corrective action and preventive action. These two aspects of CAPA have traditionally been connected, but are ideally are only distantly related. Here is the main difference between the two:
- Corrective Action: Elimination of the cause or causes of an existing nonconformity or undesirable situation in order to prevent recurrence.
- Preventive Action: Identification and elimination of the cause(s) of potential nonconformities in order to prevent occurrence.
In standards such as ISO 9000 and FDA 21 CFR 820, the description of preventive action follows directly after the description of corrective action, which has led to the misconception that the two processes must work together in series. In fact, they are separate, and preventive action ideally precedes corrective action to prevent or avoid the need for corrective action.
CAPA processes are used particularly in food processing, medical device development and manufacturing, and pharmaceuticals. FDA 21 CFR 820 is the quality system regulation that requires corrective and preventive procedures to be documented in medical device manufacturing facilities.
Corrective and preventive actions also have a place in the quality management process as defined in the Project Management Book of Knowledge (PMBOK). Corrective and preventive action is also considered a tool within Six Sigma for understanding regular business operations. CAPA has strong parallels with Design for Six Sigma (DFSS), used to design new products or redesign existing products. The analytical aspects of both corrective and preventive actions also harken back to PDCA. The component of preventive action that encourages documentation and company education on innovations and lessons learned is similar to Yokaten in lean manufacturing.
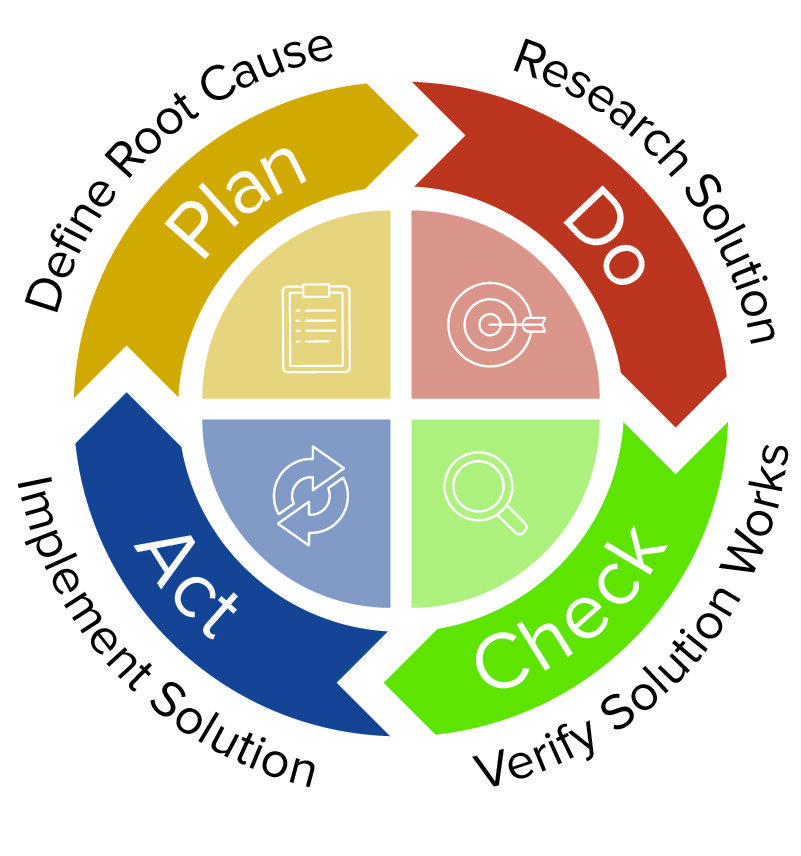
What Is the Difference between Corrective Action and Preventive Action?
Corrective is reactive. Preventive is proactive. Although these two actions use similar processes and some of the same analytical tools, they are not necessarily used together.
What Is the Meaning of Corrective Action?
Corrective action involves the identification, documentation, and elimination of the root cause of a nonconformity or problem to prevent the problem from recurring. Corrective actions are taken under more intense consideration than corrections (which address immediate issues), and you typically enact corrective actions over a slightly longer time period to prevent recurrence. For example, if you put a bucket under a leaking garburator, that’s a correction. If you inspect the entire sink and drain, learn that the unit repeatedly leaks and blocks because of a damaged seal and joint, and then remove and replace the garburator with an effective garburator that will not leak or clog, that’s a corrective action.
Most corrective action procedures use a variation of 8D problem solving. The following are some of the types of steps in a corrective or problem solving process:
- Promptly identify and document the problem. Use 5 Why questioning to acquire details and determine if this is an isolated event or if it is significant and has the potential to recur. Reporters may indicate that the problem is pervasive, but it may be limited. When documented, quality events should be reported to management. The 5 Why template below will help you identify the root cause of a problem.
Download 5 Whys Template
Excel | Word | Smartsheet
- Implement a correction or containment or temporary repair. This may include removing a defective item from production.
- Find the cause of the issue. Use 5 Whys to help pinpoint a problem statement. Use an Affinity or Ishikawa (fishbone) diagram to help determine the root cause. You can use the free Ishikawa diagram template below to get started.
Download Fishbone Diagram Template - Excel
- Determine the solution that will prevent a recurrence. Solutions can include new parts, process changes, and even system changes.
- Implement the corrective action and ensure that everything is documented.
- Verify that the action continues to be effective and that the problem does not recur. Document the evidence of continued success.
Risk-Based Prioritization
Along with the defined corrective action procedures, a predefined risk-based prioritization eliminates small nonconformities that teams can solve when they discover problems without initiating corrective procedures. Weight matrices help with these questions — the criteria often include frequency (frequently, occasionally, or rarely) and impact (negligible, critical, or even resulted in injury or death). Safety usually trumps frequency in action response.

What Is Preventive Action in ISO?
Until the release of ISO 9000:2015, preventive action was one of the few mandatory procedures required by ISO 9001. Companies were also required to keep records on nonconformities and preventive actions taken. However, some practitioners considered the standard to be unclear regarding where to look for potential problems. Pundits explained that preventive actions concerned risk and directed practitioners to ISO 9004, “managing for the sustained success of an organization — a quality management approach,” which was considered a pointer to what preventive actions should address.
ISO 9000:2015 eliminates the requirement for predefined procedures for both corrective action and preventive action. In fact, preventive action is now considered a part of good planning and risk management. It fully incorporates the notion that prevention comes first and eliminates problems and, thereby, the need for corrective action. As of 9001:2015, you simply document what happened and how you fixed it. Following are the essential CAPA-related definitions:
- Correction or Immediate Action: This eliminates the immediate problem. It doesn’t eliminate the issue permanently, but it allows a process or work to continue. In PMBOK, correction is also known as defect repair. An example of this process is mopping up water and adding a bucket under the leak.
- Corrective Action: This eliminates the cause of the nonconformity and prevents repetition. Corrective actions move products, procedures, processes, and projects back to baselines. On a large scale, corrective action is necessary if a project moves away from the project management golden triangle of budget, schedule, and quality. An example of corrective action would be investigating the sink, drain, and supply system, learning that the garburator backed up twice before, replacing the garburator, and confirming that nothing leaks.
- Preventive Action: This prevents potential occurrences. Preventive action determines what in a project might veer away from the project management golden triangle of budget, schedule, or quality. An example of this process would be checking the other garburator in another sink as well as U traps in two other sinks for existing problems and asking if any parts should be replaced now before they fail.
The following are tools that you can use to analyze risk or potential problems:
- Hazard and Operability Study (HAZOP): A HAZOP is a sequenced and methodical study of a process that is in development or in operation. The HAZOP seeks to identify problems that may represent risks to personnel or equipment.
- Failure Modes and Effects Analysis (FMEA): FMEA is a step-by-step approach for identifying all possible failures in a design, a manufacturing or assembly process, or a product or service. Failure modes means the ways (or modes) in which something might fail.
- Fault Tree Analysis (FTA): FTA is commonly used in safety and reliability engineering to break a system into subsystems to understand where problems may occur. FTA is often used in pharmaceutical development and manufacturing.

What Is a Preventive Action Process?
A preventive action process, in addition to including a specific preventive action plan to mitigate potential problems, also comprises the implementation of controls to ensure that any preventive measures continue to work. Preventive action means identifying not only potential problems, but also opportunities for improvement. With changes that are enacted through a preventive action process, controls should be included to prevent and check for possible nonconformities.
What Is an Example of Preventive Control?
A preventive control, also known as an internal control, serves to reduce the chances of problems and nonconformities that occur. Although many fields employ them, preventive controls form a particularly important part of food preparation quality control. Under the FDA’s Food Safety Modernization Act (FSMA), for example, certain food preparation facilities must write a food safety plan, which begins with a hazard analysis and addresses specific types of controls. For instance, among other preventive controls, food preparation must include allergen tracking, and packaging must be labelled appropriately. However, preventive controls can be as simple as employees washing their hands and segregating utensils used for raw foods. The FSMA lists corrections and corrective actions as a management aspect of preventive controls to be implemented in quality events, such as when a deviation from a preventive control occurs. The practice of preventive controls for food safety even has its own professional support group, the Food Safety Preventive Controls Alliance (FSPCA).
Approaches to Preventive Analysis and Actions
One complaint with regard to CAPA in ISO 9000 was that it was vague about how possible problems might be found. So, where do you look for for possible problems? Think about what could go wrong.
According to Conover, “It’s when you’re in production. That’s when all the unanticipated risks typically occur. It’s where you think, ‘Oh my gosh, we didn’t anticipate that this would happen.’” He says that’s why the analogy to a human immune system is strong: “Your immune system reacts to antigens that are unknown or toxic to the body. Once the antigens are discovered, [your immune system] applies antibodies. In the future, in some cases, T-cells will selectively recognize previously known antigens and minimize any effect. Just like with CAPA, those things that happen in pre-production, production, or post-production should have corrective and/or preventive actions that ensure that they do not occur or recur,” he emphasizes.
Teams may find potential problems in internal or external data sources. Internal data sources may include process control data, trend analysis, the results of proficiency testing, or internal audits. External data includes customer complaints, service reports, and even data for similar products produced by other companies.
Risk analysis should be done when you adopt new tools or software. Preventive actions may also be sparked through the results of regularly assessing employees and then deciding if more or revised training is required. Risk analysis can help target costs, so the exercise doesn’t appear to be a waste of money when problems don’t occur. FMEA is one tool to view potential problems.
Here are some examples of preventive actions:
- Creating disaster recovery plans for hazards and unexpected situations throughout your facility
- Implementing or updating safety and security policies
- Creating checklists
- Implementing lean practices to reduce waste that can contribute to problems and nonconformities
- Implementing and following preventive maintenance plans to ensure that equipment performs efficiently, effectively, and safely
Some experts still think that preventive actions follow the experience of corrective actions. This sensible approach focuses on capturing the experience for the future, including tagging keywords from the action report in databases and updating documents, such as FMEA, requirements documents, and procedures.
What Is 21 CFR 820?
21 CFR 820 stands for Title 21 Code of Federal Regulations 820. Title 21 governs food and drugs for the FDA. CFR 820 is the quality system regulation that applies to manufacturers of medical devices. 21 CFR 820 borrows heavily from ISO 9000: It requires facilities to create and maintain procedures for implementing corrective and preventive actions so that employees can address problems with devices currently or imminently on the market.
A well-defined CAPA process, or subprocess, as the FDA calls it, provides structure for accomplishing three things:
- Gathering and analyzing information to find existing and potential problems and nonconformities
- Observing quality issues and applying effective corrective or preventive actions as needed
- Ensuring that corrective and preventive actions have been effective
In the FDA’s view, a CAPA process also provides a structure for communicating CAPA activities to employees, reporting to management, and documenting activities for review and future improvement purposes.
The CAPA concept is also integral to the Current Good Manufacturing Process (cGMP), an approach advocated by the FDA. CAPA may be applied to a variety of aspects of product development, such as design, production, product testing, and post-market use. CAPA may also be applied in product packaging, distribution, and shipping.
Questions in a CAPA Inspection
When the FDA inspects your facility, it may seek to ensure that you have documented CAPA procedures and make sure that they contain certain elements. For example, your records should show that you could find the root cause of problems and that you are tracking trends to ensure you avoid future problems or recurrences of problems. Investigators also look for evidence that you tested CAPA actions to ensure effectiveness before you implemented them. Moreover, the FDA wants to verify that the data source and statistical process methods you employed were sufficient for the task.
What Is a CAPA Report?
A CAPA report provides a consistent vehicle for recording defects and issues as well as the method of their correction. Usual details include where the problem occurred, the customer’s name and address, the details of the problem, whether there was a product breakdown, whether there was an injury, and so on. The report also states what immediate action or correction was taken. The report may walk you through the process, suggest tools for the root cause analysis (such as 5 Whys and cause and effect analysis), and provide room to record analysis results. It may also provide guidance on how to route the report depending on the outcome of the analysis.
You can use the free template below to create a CAPA report.
Download CAPA Report Template
Excel | Word | Smartsheet
Misconceptions About CAPA
Although regulations may require organizations to document CAPA processes and follow them during quality events, and tons of web content exists concerning how to implement CAPA, employees and managers often harbor the following doubts and misconceptions about CAPA:
- It’s a Punishment because Something Has Gone Wrong: For the health of a company, someone has to implement the CAPA process. Training a dedicated CAPA team can help to depersonalize CAPA assignments.
- It’s Extra Work: “When everybody has a part-time job of being an investigator, typically nothing changes,” says Conover. He gives the example, “‘You’re a QE and, by the way, in your spare time, which you don’t have, you’re an investigator.’” In that scenario, he says, the extra work remains undone. The solution is to establish a review board with people trained in appropriate roles so that CAPA becomes a regular responsibility.
- Training Is Too Expensive: Management often complains that neither budgets nor schedules offer resources for training employees in the efficient execution of CAPA. You can save money by having the process in place. You should also ask yourself how much it costs to have a product recalled. “Here are our biggest comments: We don’t have time to train. We don’t have the money to train. There's no budget. I can’t pull people off the floor for a day and a half,” says Conover. The bottom line? “Organizations that train are the ones that sustain, and those that don’t won’t survive beyond five, 10, or 15 years,” he concludes.
Software Solutions for CAPA
The many details and documents necessary for a good quality management system are represented in CAPA. Although software can’t make up for a poor CAPA procedure or lack of follow through, a strong platform can help track the many updates that should be added to assorted documents. Software can also support documentation and audit trail requirements for the FDA’s GMP, GLP (good laboratory practice), and GCP (good clinical practice). With a web-based system, authorized users can access a central repository to get all the documents and information they require. Online templates and automated workflows provide routing, notification, and electronic approval. Finally, software can help with analytics and reporting.
Improve CAPA Procedures with Work Management in Smartsheet
Empower your people to go above and beyond with a flexible platform designed to match the needs of your team — and adapt as those needs change.
The Smartsheet platform makes it easy to plan, capture, manage, and report on work from anywhere, helping your team be more effective and get more done. Report on key metrics and get real-time visibility into work as it happens with roll-up reports, dashboards, and automated workflows built to keep your team connected and informed.
When teams have clarity into the work getting done, there’s no telling how much more they can accomplish in the same amount of time. Try Smartsheet for free, today.