What Is the Research Protocol?
All clinical research starts with the research protocol, a document that details all aspects of the trial: its background, rationale, objectives, design, methodology, statistical analysis plan, and organization. With the protocol, you can make sure you protect the participants and collect the data. Using protocol templates, you can start thinking through what you need to meet compliance standards with the Food and Drug Administration (FDA) and clinical study best practices.

Download Research Protocol Template - Word
The full research protocol includes the following sections and topics:
- Title Pages: These pages provide general information about the protocol, including name, number, version number and date, trial phase, investigational product name, investigational new drug (IND) number, sponsor (or principal investigator in academia), funding organization, medical monitor, and coordinating center. The pages include the principal investigator’s signature (or sponsor), as well as site-specific information, such as the agreement, and protocol details. They also detail the study team and site, particularly in the case of multiple teams and sites.
- Objectives: List the study’s primary and secondary objectives.
- Background Information: Describe the problem under study and priority. Include the medical and scientific rationale that justifies researching the problem. Include data from other studies relevant to this proposed research. Include the name and description of the proposed intervention, including the dosage, route of administration, period, and frequency of intervention.
- Study Design: Describe the methodology and how it will answer the study question. This should include the type of study, primary and secondary outcome(s), population, sample size, study location, period of enrollment and follow-up, intervention and route of administration, randomization (as necessary), and any other relevant protocol information.
- Selection and Exclusion of Subjects: Provide statements describing how the participants must meet all the inclusion and exclusion criteria, and list the criteria. Clearly define the study population. For example, list the demographic criteria, required laboratory data, any prior therapies allowed or disallowed, ability to understand and meet all study requirements, if contraception is necessary, exclusion criteria such as specific health status, use of excluded drugs, cancer status, and chemical dependency status.
- Study Enrollment Procedures: Describe the methods and procedures for identifying and enrolling subjects, how they are documented, how consent is obtained, and any randomization procedures.
- Study Intervention, Duration, and Route of Administration: This section should describe each intervention and duration, as well as how each is administered. List expected adverse effects and dose escalation, if applicable. Discuss how the intervention is acquired, stored, and disposed of, as well as documentation for intervention accountability. In addition, note the medications restricted, allowed, and required, along with the extent to which these medications are tracked and documented.
- Study Procedures: This section includes a study evaluation schedule (presented as a chart) and explanations of the required assessments, what each period is, and any special considerations or instructions necessary. These should match what is available in the column headers of the chart above, and they should include information on the screening or baseline assessments, randomization, blinding, follow-up visits, and final assessments.
- Safety Assessment: List any expected adverse events, and how these could be managed. Mention any toxicities seen in earlier IND studies here. Also, include safety measures as identified in laboratory findings, methods and timing for safety parameters based on the risk profile, definitions for adverse events (AE) and serious adverse events (SAE) and laboratory values used to identify their possibility, timeframes for reporting and collecting information on AEs and SAEs, the reporting system, how you will follow up on AEs, and the specific guidelines for independent monitoring.
- Intervention Discontinuation: List criteria for intervention discontinuation and how you could meet them. Also list possible reasons for discontinuation, any modifications to the schedule should it be discontinued, duration of follow-up, any temporary discontinuation criteria, or any evaluations should participants be temporarily or permanently discontinued from the study.
- Statistical and Analytical Considerations: Include primary and secondary statistical hypotheses, why you chose the study design, the primary and secondary outcome measures, and the validity and reliability of these measures. Also discuss sample size and randomization, treatment assignment procedures, how you define the population, any interim analyses, primary and secondary outcome analyses, the statistical methods you use to consider any necessary intervention effect between groups, and if necessary, the expected positive within group correlations among different study arms.
- Data Collection: Detail how you will gather the data, the required forms, how to keep these forms confidential, and what source data to expect. Note site responsibility for data collection and management, and (if necessary) the responsibilities of the coordinating center.
- Quality Assurance: Describe training for study staff, whether there is a control committee and their required practices, any quality control metrics, how you will identify and document protocol deviations, how you will assure protocol compliance, and the schedule for reviews. If you have a manual of procedures (MOP), reference it here.
- Participants Rights: Include references to the Institutional Review Board (IRB) requirements, informed consent documents, procedures for participant confidentiality, and study discontinuation requirements.
- Committees: List any committees associated with the study, along with their roles.
- Publication: Outline the requirements and procedures for publication.
- References: List any citations referenced in this protocol.
- Supplements/Appendices: Include any additional documentation.
To track every aspect of the proposed research for each participant, create a case report form (CRF) that you can use in both paper and electronic formats. With CRFs, you can collect and analyze data for analysis, and then generate a conclusion for your study. For more information on the distinct phases of clinical trials, see “Understanding the Phases of Clinical Trials.”
Concept Protocol Template
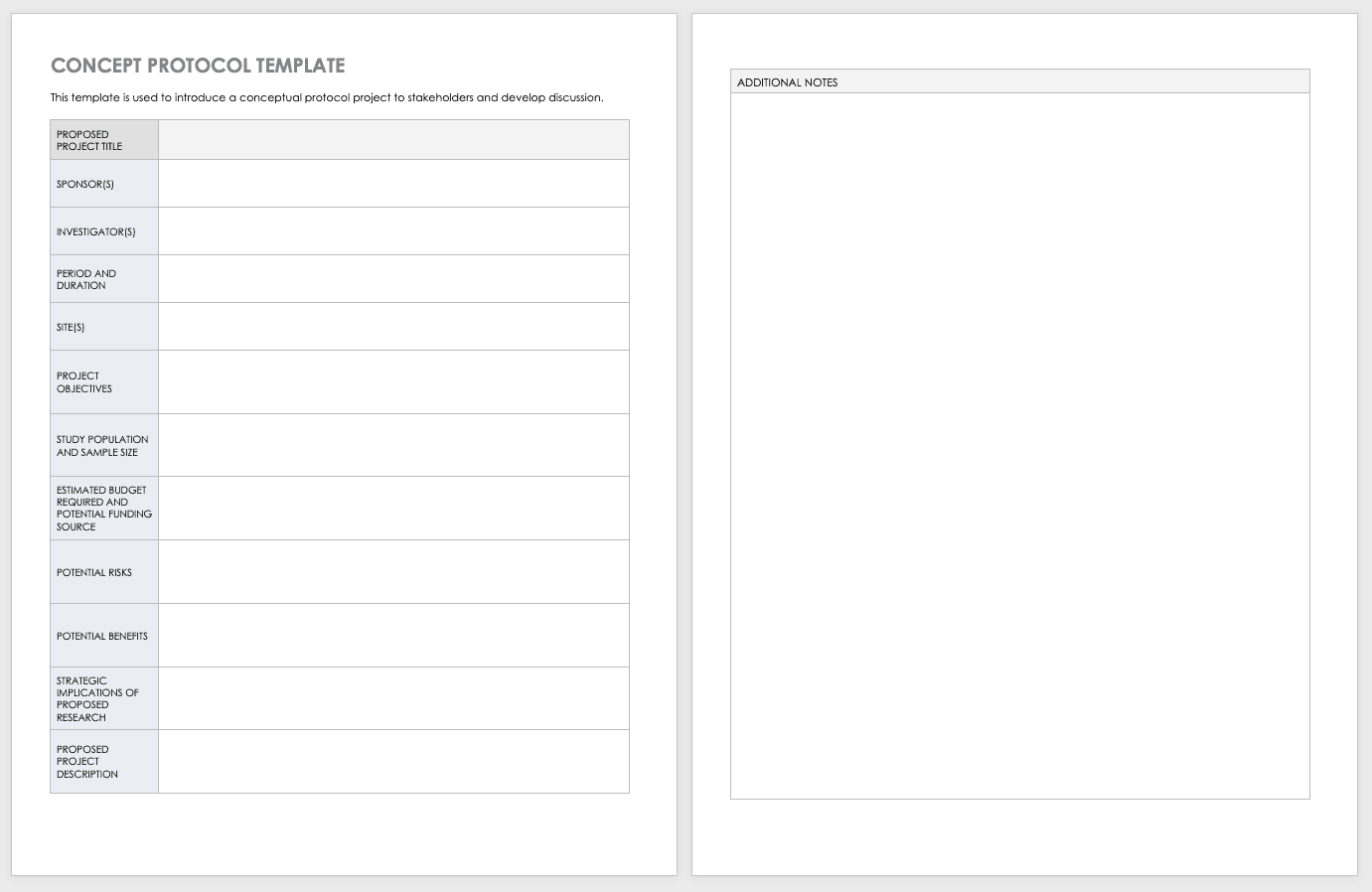
Phase 1 Clinical Trial Protocol Template

For nonclinical research or clinical trials that are Phase 0 or Phase 1, use this free template. Phase 1 or nonclinical trials do not require the same amount of detail as a full study protocol.
Download Phase 1 Clinical Trial Protocol Template - Word
Research Compliance Templates
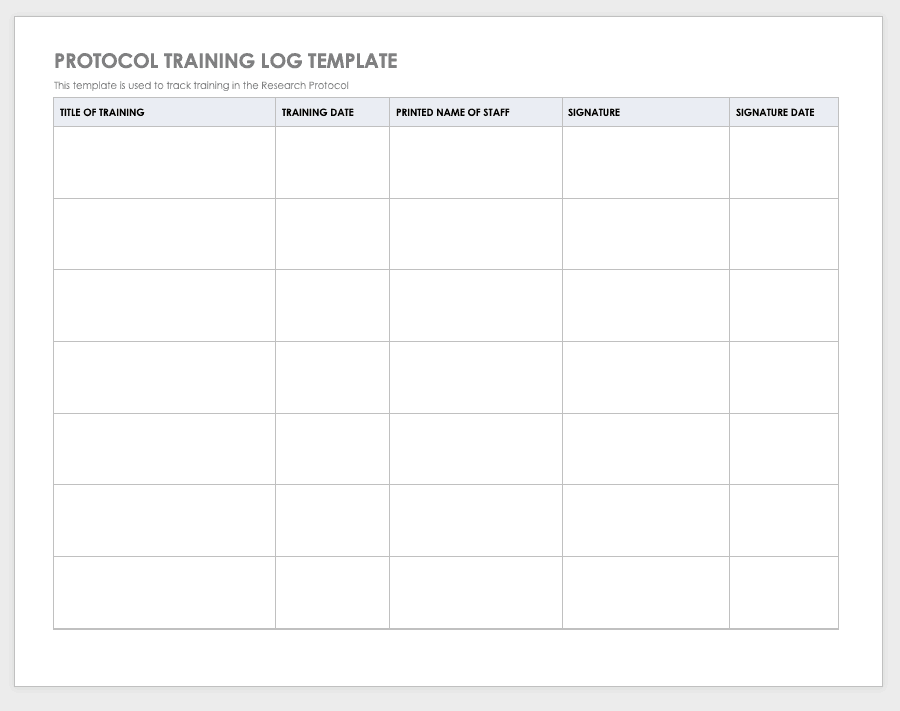
By training staff members on the research protocol, you’ll help them meet compliance standards and understand the purpose and details of the study. Use a training log to record all training that the site study staff completes, signing the log entry for verification.
Download Protocol Training Log Template
Excel | Word | PDF | Smartsheet
Protocol Deviation Template
Protocol deviations are inadvertent or unplanned changes or noncompliance with the research protocol. These events do not increase risk or decrease benefit, nor do they impinge on participants’ safety or rights. They do not compromise study data, but you should capture the deviation for reference.
Download Protocol Deviation Log Template
Delegation of Authority Log Template
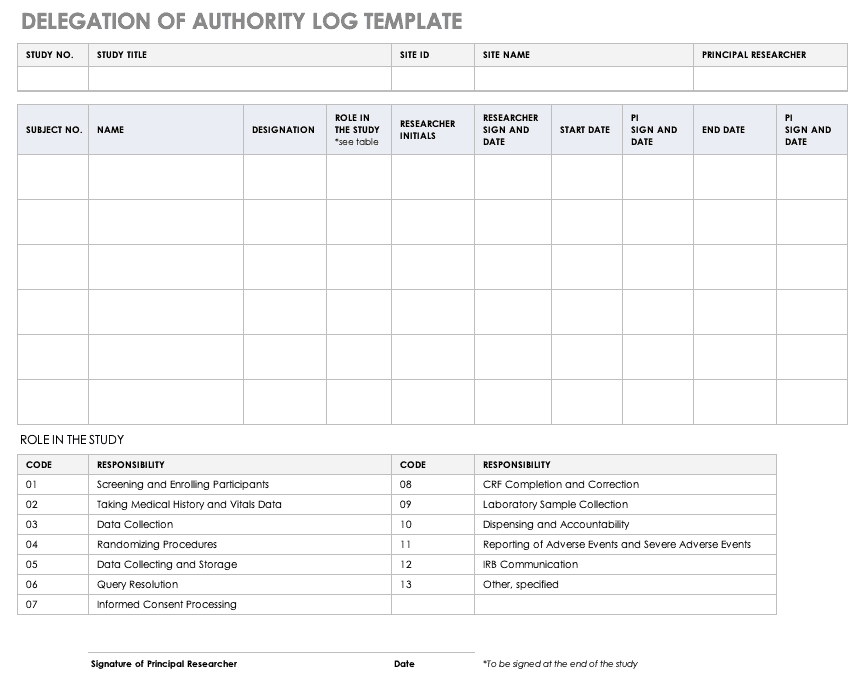
Once you’ve trained your staff and figured out their roles and responsibilities, the principal investigator must delegate authority. The delegation of authority log should be filled out and signed prior to the study’s start.
Download Delegation of Authority Log Template
Excel | Word | PDF | Smartsheet
Site Selection Visit Form Template
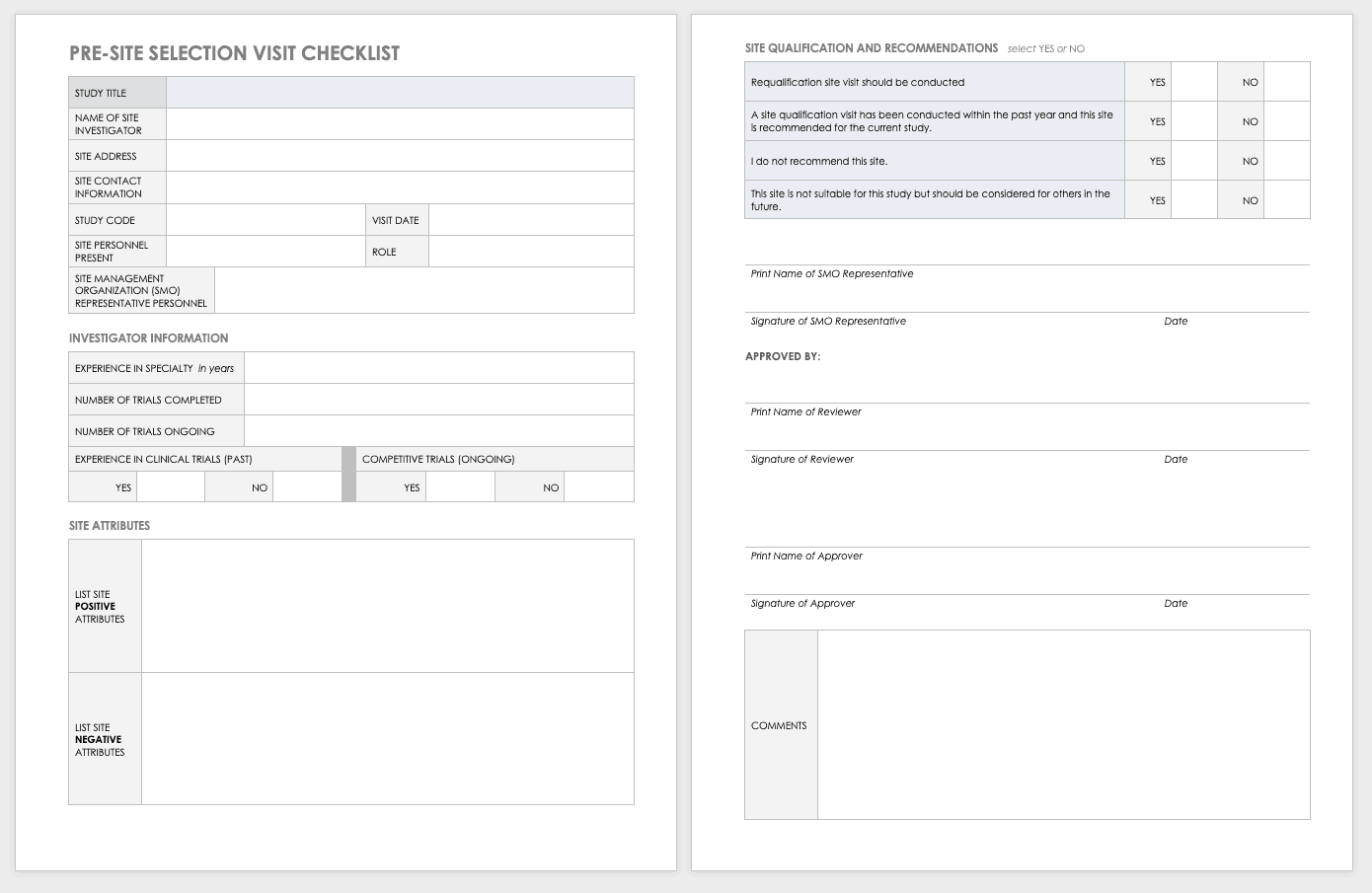
The sponsor must perform a site visit to determine its suitability as part of a multisite study. This means taking a tour to determine whether the site has the capabilities to meet the sponsor’s goals.
Download Site Selection Visit Form Template
Word | PDF | Smartsheet

Project Management for Clinical Trials, Practices, Templates, and Documents
Clinical trials are big projects. If the organization is not used to planning and wants to conduct clinical research, it must hire a project manager and work with senior leadership to introduce planning into the organization.
Together, they should develop the main goals and define their limits and the terms of success. They should set out a strategy for which tasks and sets of tasks to perform and in what manner. Test any planning tools or software before the trials start. When possible, use templates to ensure consistency and best practices.
Once the trial starts, evaluate your systems with standardized metrics. The project manager can track study deviations and apply corrective actions. Use the lessons learned from past and current projects to help guide future projects. Employing consistent tools gives you the opportunity to draw from a reservoir of data.
Clinical research can cost billions of dollars and years of time, resources, and effort. As
such, project management best practices and methodologies are critical to the success of a clinical trial, according to experts.
Many software systems are available to manage clinical trials. When very specialized, these are referred to as clinical trial management systems (CTMSs). However, other platforms can also manage clinical trials and may already be embedded with your information technology. Regardless of the platform you use, you should have full project management functionality, such as planning and reporting modules, as well as the ability to track participant contact information, deadlines, and milestones.
You may want to consider the following project management documents for your clinical research.
Project Management Plan (PMP) for Clinical Trials
A PMP delineates and acts as an agreed-upon document of scope, responsibilities, and guidance. You can use it throughout the project to help stay on track. Every clinical trial has difficult milestones, but a good project management plan can help you sidestep some of the regular issues.
You have many PMP software platforms to choose from, but regardless of your ultimate decision, your PMP must focus on protocol adherence, subject care, and service quality, along with how to achieve each standard. Here are the sections you should include in your PMP for a clinical trial:
- Project Objectives: This is an outline of the research objectives for the study, your quantifying standards, and your goals.
- Background and Strategic Context: By documenting background and context, you establish a foundation for decisions and discussion to follow.
- Study Governance: The governance covers the roles and responsibilities in the project, encouraging open communication, sharing, and accountability.
- Stakeholder Management Plan: This plan details how the staff and investigators will collaborate and effectively communication with stakeholders. This could include (as per the roles and responsibilities) regular emails, newsletters, consultation, oversight, training, and documentation.
- Scope: This document delineates assumptions, constraints, and deliverables (and their expected dates).
- Project Risk Assessment: This document helps you prepare for risks and decide on the risk profile.
Clinical Research Project Activity List
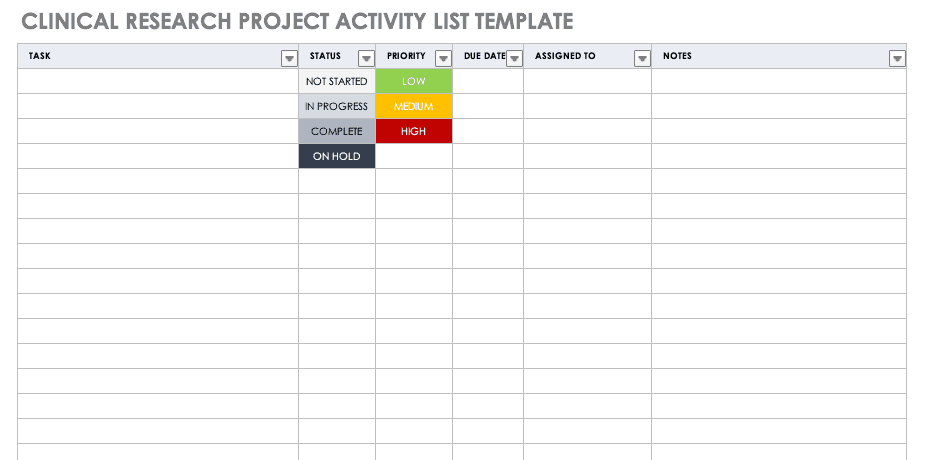
A project activity list is an itemized documentation of all the activities scheduled as part of the project. This list should be very detailed, including the status and priority of the task, when it is due, and to whom it is assigned.
Download Clinical Research Project Activity List Template
Clinical Trial Timeline Template
A timeline enables you and your staff to track each major portion or milestone of your clinical trial. Your timeline should include these steps:
- Choose Research Questions and Study Design: Research always begins with questions. Your research question will determine how you design your study.
- Choose Outcomes: The outcomes for any trial are dependent on many factors, including scope, health conditions under study, target population, type of intervention. One resource to help develop outcomes is Core Outcome Measures in Effectiveness Trials (COMET). This database details core outcome sets for comparison in clinical trials.
- Prospectively Register the Trial: Whether you are working through the FDA, World Health Organization (WHO), or another national agency, study transparency is critical. Prospective registration of trials is recommended. One resource for registration is the ISRCTN registry.
- Obtain Ethics Approval: Any trial involving human participants must go through an ethics review to safeguard the subjects’ rights, safety, well-being, and dignity. There are many options for institutional review, including through a university or a private or governmental organization. Without this step, research cannot commence.
- Prospectively Publish Protocol and Analysis Plan: Before a clinical trial, you must complete some pilot research. When you publish the research leading up to a clinical trial, along with the protocol and analysis for the trial itself, you increase transparency and accountability of the research.
- Planning for the Trial and Data Management: Many clinical research professionals recommend including patients in the planning phase of clinical trials, at least as stakeholders to review the plan. By completing the plan early and allowing potential participants to review it, you help improve recruitment and retention during the trial.
- Recruitment and Retention: Recruitment is getting the right people to take part in your trial, and retention is about keeping their interest and trust. A source of unending frustration for researchers, recruitment and retention can make or break a trial.
- Identify and Manage Trial Sites and Staff: This process is not as straightforward as it is often thought to be. Study coordinators must use feasibility checklists to choose sites and figure out how to get bring on staff who have the bandwidth to recruit for the study.
- Data Collection: The methods for collecting data are critical to any study. Advance planning and structure help you stay organized, comprehensive, and transparent so that your study can have a seamless analysis and solid conclusions.
- Data analysis: Flaws in analysis can generate poor, biased, or erroneous outcomes. In advance, researchers should consider patient blinding, randomization procedures, and sequence generation.
- Findings dissemination: Some researchers recommend threading all research on a trial topic. One resource for this is CrossRef, a database that links similar research. Regardless, the point of research is to capitalize on scientific progress and move it along. By having a plan to disseminate your results, you ensure that others capitalize on your research and move the knowledge forward.
Use this free template to develop your own clinical trial timeline. Add your own steps, milestones, and dates for a comprehensive, expansive view.
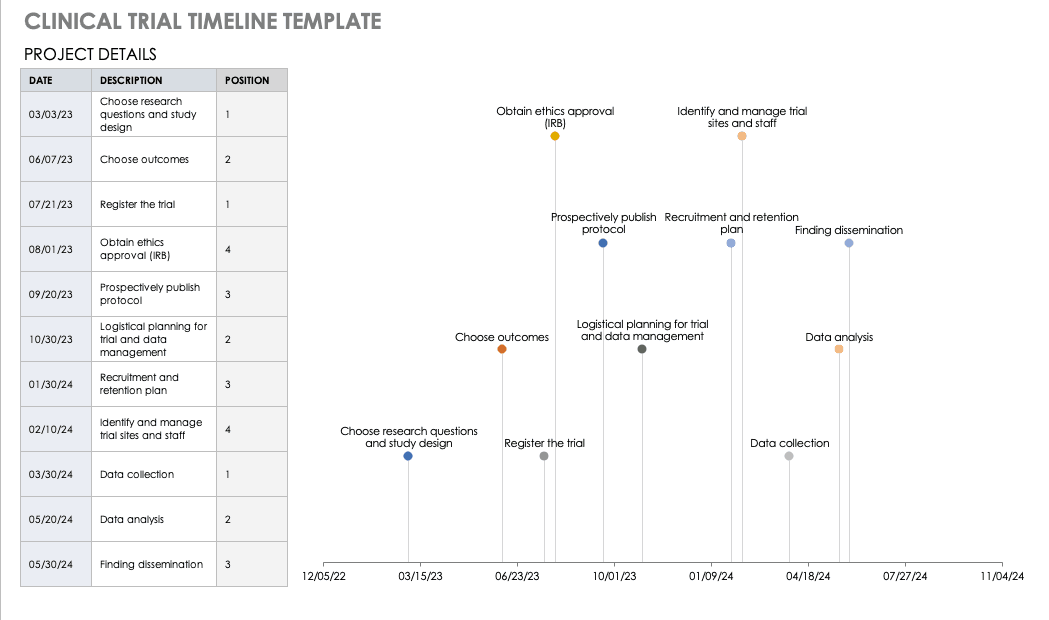
Download Clinical Trial Timeline Template
For a different perspective, add your project details to this free template so you can view your timeline visually.
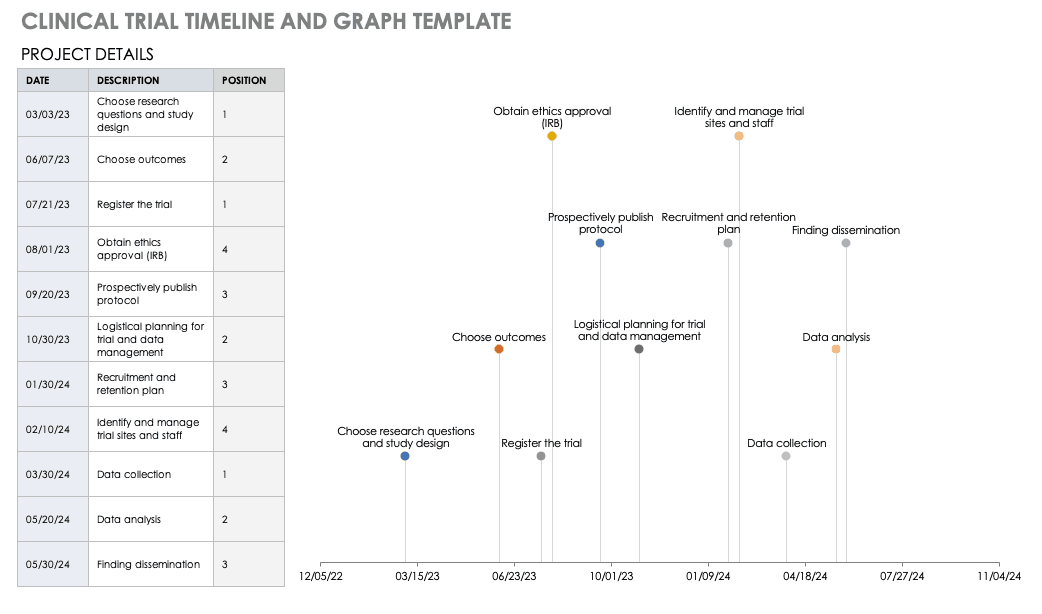
Download Trial Timeline and Graph Template
Microsoft Project Management for Clinical Trials
First released in 1985, Project is a well-respected Microsoft product for project management. Microsoft Project was not traditionally available as a part of Office Suites, a package of programs for professionals and professional organizations. However, Microsoft recently included it as a part of the Windows 2016 suite.
Microsoft Project Management has the following features:
- Built-in templates
- Project portfolio management
- IT management
- Presentations
- Out-of-the-box reports
- Multiple timelines
- Real-time reporting
- Dependency management
- Priority assignment
- Lean management
- Gantt charts/project mapping
- Calendar views
- Setting baselines/KPIs
- Project budgeting
- Issue tracking
- Task creation
- Resource management
- Cloud access
Microsoft Project has built-in templates that you can apply to clinical trial management.
Microsoft SharePoint for Clinical Trials
SharePoint is a collaboration platform that is integrated with Microsoft Office. SharePoint manages and stores documents, and it enables multiple users to access the documents via their own site or a standardized Microsoft site. A subscription to Microsoft Office 365’s SharePoint does not require a server, but customization options are limited; the flexible authentication and authorization systems are built in.
SharePoint Server, available in Standard or Enterprise versions, can be developed as either
virtual or hosted services in a business’s IT department. SharePoint Server enables the organization to control the SharePoint features available to staff, and you can scale it to meet different numbers of users.
Windows SharePoint Services 3.0 is a Microsoft-hosted version that comes with Microsoft Office. Microsoft provides a template in SharePoint for Clinical Trials: Clinical Trial Initiation and Management application template for Windows SharePoint Services 3.0. You can download and add this template to your SharePoint Services, which enables you to create the following:
- Clinical Trial Protocols: This includes the objectives, study design, project plan, subject selection, and budget.
- Protocol Documents: This includes additional documents relative to your study.
- Calendar: Track milestones in the project.
- Threaded Document Discussions: Team members can start and track discussions within documents.
- Task Creation and Assignment: You can create and assign tasks to users, who receive email notifications.
- Archiving: You can move documents or groups of documents to archive status, keeping them but not making them visible.
The clinical trial template has site lists of libraries for clinical trial protocols, protocol documents, announcements, calendars, issues, tasks, and document discussions. These can be further customized with different versions of SharePoint. To download this template, you will need access to SharePoint Server 3.0.
Clinical Research Budget Plan Template
In many instances, you set the clinical trial budget after much negotiation with a sponsor. Other times, you need to build a budget before the sponsor is even on board, as a way to convince them of the project’s feasibility. The key cost drivers for any clinical research project are the following:
- Patient Grants: These include the costs for screening failures, baseline patient measurements, and procedural costs.
- Site Costs: This covers any expenses associated with the site, such as start-up fees, IRB fees, storage fees, and site management costs.
- Non-Patient Costs: This includes consultation fees, monitoring board fees, and any medical device costs.
- Labor Costs: You must account for all the staff required for the project and their full-time equivalency (FTE).
- Site Management: These costs include pre-study visits, initiation fees, monitoring, and close-out fees.
- Miscellaneous: These include investigator meetings, any technology needs, and ad hoc travel.
- Unexpected Costs: These are costs resulting from protocol amendments, value added tax (VAT), delays, and inflation.
Before you start putting together your research budget, you must gather the following:
- Schedule of assessments from the protocol
- Standard institutional fees from your institution, if applicable
- Evaluation and procedural costs
- Site costs
- Staff allocation and their hourly rates
- Indirect cost rate
- Subject compensation costs
- Data storage fee estimate
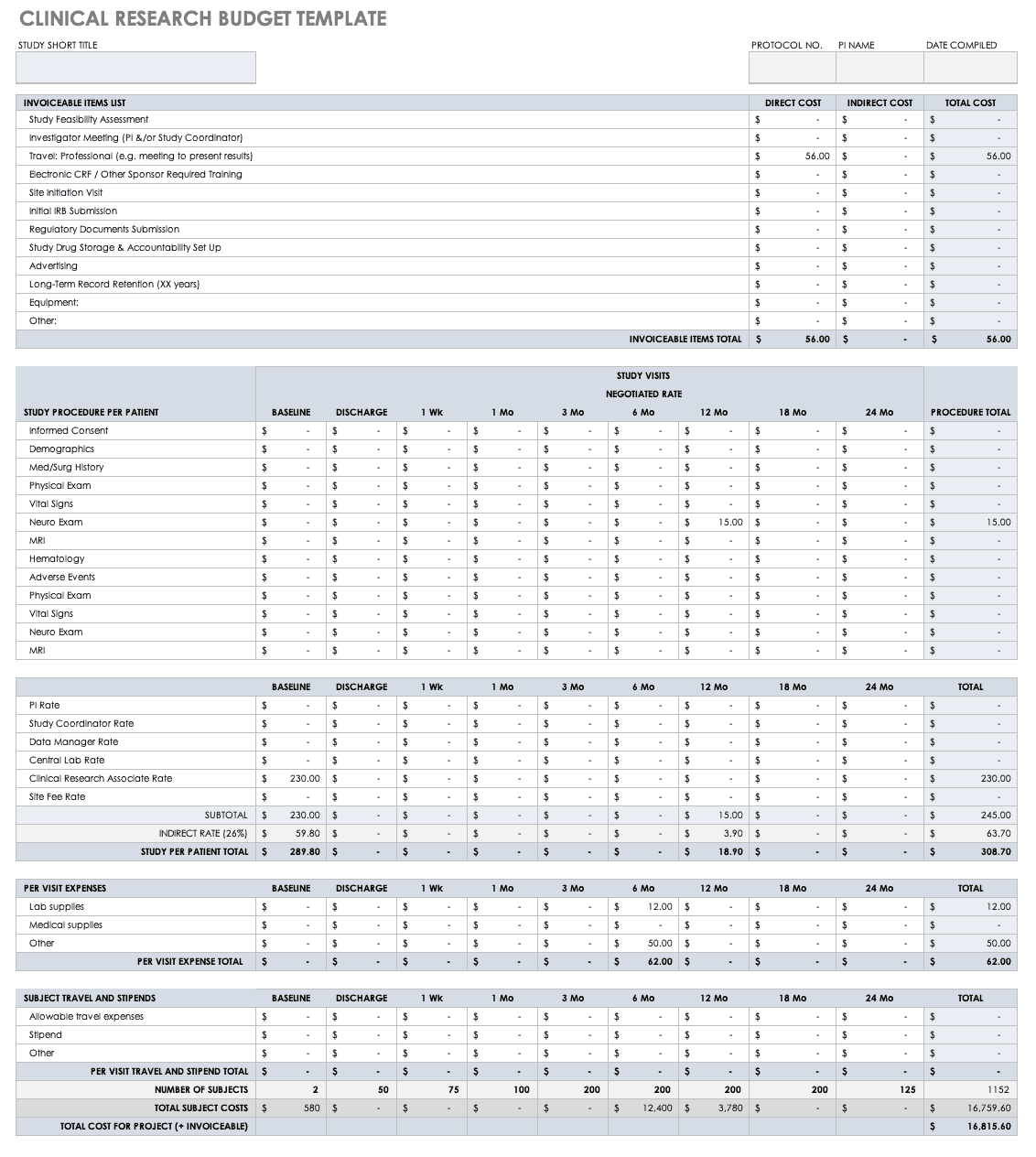
Put together your own clinical trial budget with this free clinical research budget template.
Download Clinical Research Budget Template - Excel
Clinical Research Tracking Log Templates
Clinical research requires scrupulous planning, a well-developed team, regulatory adherence, and above all, excellent documentation. It is therefore critical for clinical trial project managers to have a completed scope of work and to develop all the forms and templates before the trial begins. Some of these documents are for planning, and some, like those included below, are for operational purposes.
Regulatory Binder Checklist

Strong clinical practice thrives with a regulatory binder checklist. This checklist keeps track of all paper versions of essential regulatory study documents. Each document should also include any electronic locations. This document should be regularly updated, customized for unique studies, and stored in reverse chronological order.
Download Regulatory Binder Checklist
Clinical Study Document Tracking Log
It is important to not only track all paperwork related to a clinical trial, but also be able to locate it easily between various staff and sites. A clinical trial document tracking log can help you keep a written trail of the documents and when they were submitted and approved. You should also keep copies of the documents with the log. Use this free template to develop your own clinical study document tracking log. You can also adapt the log for specific correspondence, such as documents relating to FDA or IRB submissions, but it should not be mixed with regulatory documentation.
Download Clinical Study Document Tracking Log
Excel | Word | PDF | Smartsheet
Data and Safety Monitoring Plan (DSMP) Template
Before you can undertake a study, you must develop a DSMP for how to keep participants safe and how to secure data and ensure accuracy. The DSMP has several sections:
- The study purpose
- An adherence statement
- Any protocol amendments
- Multisite agreements
- A plan for subject privacy
- Confidentiality during adverse event reporting
- Expected risks
- Adverse events, unanticipated problems, and serious adverse events: how they are defined, their relation to the study, expectations, severity grading, and reporting procedures in single-site and multisite trials, and whether they are IND or non-IND studies
- Events of special interest
- Pregnancy reporting
- Rules to halt the study for participants
- Quality control and quality assurance
- Subject accrual and compliance
- Sample size justification
- Stoppage rules
- Monitoring committee designation
- Safety review plan
- Study report plan for independent monitors
- Plan to submit reports from onsite monitoring and audits
- Data handling and record keeping
- Informed consent
- Reporting changes in study status

Create your own data and safety monitoring plan using this free template. It lays out each section so you can specify them for your research. The principal investigator should sign and date this document once it is complete so that it may be filed.
Download Data and Safety Monitoring Plan Template - Word
Research Communication Plan Template
A communication plan should describe how you will converse with internal and external stakeholders during your project. Your communication plan should include a brief overview of your project and a breakdown of the messages you need to get out. You should adapt the messages for different audiences and define who will deliver these messages. The messages should include the following:
- The purpose and benefits of the research
- The known effectiveness of the intervention, or (if the intervention is under study) the disclosure that the effectiveness is unknown
- How participants will be protected
- The risks and benefits of participating

Develop your own communication plan using this free clinical trial communication plan template. This template also includes a section for situation analysis and risk analysis that asks for inputs on strengths, weaknesses, opportunities, and threats.
Download Clinical Trial Communication Plan Template - Word
Participant Management in Clinical Trials Using Templates
A few main documents help ensure that your participants are tracked and well-cared for before and during your research study.
Enrollment Log for Clinical Trials Template
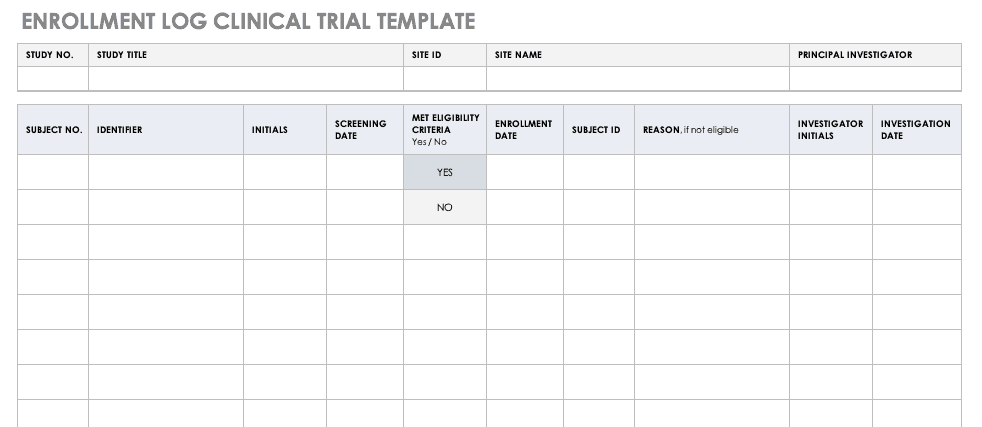
This log keeps track of everyone that has been enrolled for participation in your study. This does not mean that they have met the eligibility requirements or have been otherwise screened, but it is a record that they have signed up to be admitted.
Download Enrollment Log for Clinical Trials Template
Excel | Word | PDF | Smartsheet
Informed Consent Form Templates
Informed consent is the central tenet of ethical research with human subjects. The consent process typically involves a researcher delineating what is involved in the study, its risks and benefits, what a participant’s duties entail, and answering any questions they have. Before you perform any research, make sure the informed consent document is signed and the participant receives a copy, unless the informed consent document has been waived by an institutional review board (IRB). Federal regulations 45 CFR 46.116 govern what you must provide in the informed consent process in the United States.
To prepare informed consent documentation, researchers must do the following:
- Use plain, easily understandable language no higher than an 8th-grade reading level.
- Tailor documents to the potential population.
- Avoid technical jargon.
- Use the second or third person (you/he/she) to present study details.
- Include a statement of agreement.
- Ensure that the consent document is consistent with information in the IRB application.
These templates assist the principal investigator in the design of their informed consent forms (ICFs). You can adapt them to accommodate the details of any study and include both the information sheet and the consent form. Modify each section with the appropriate description described in italics. Use the general template for any type of research.
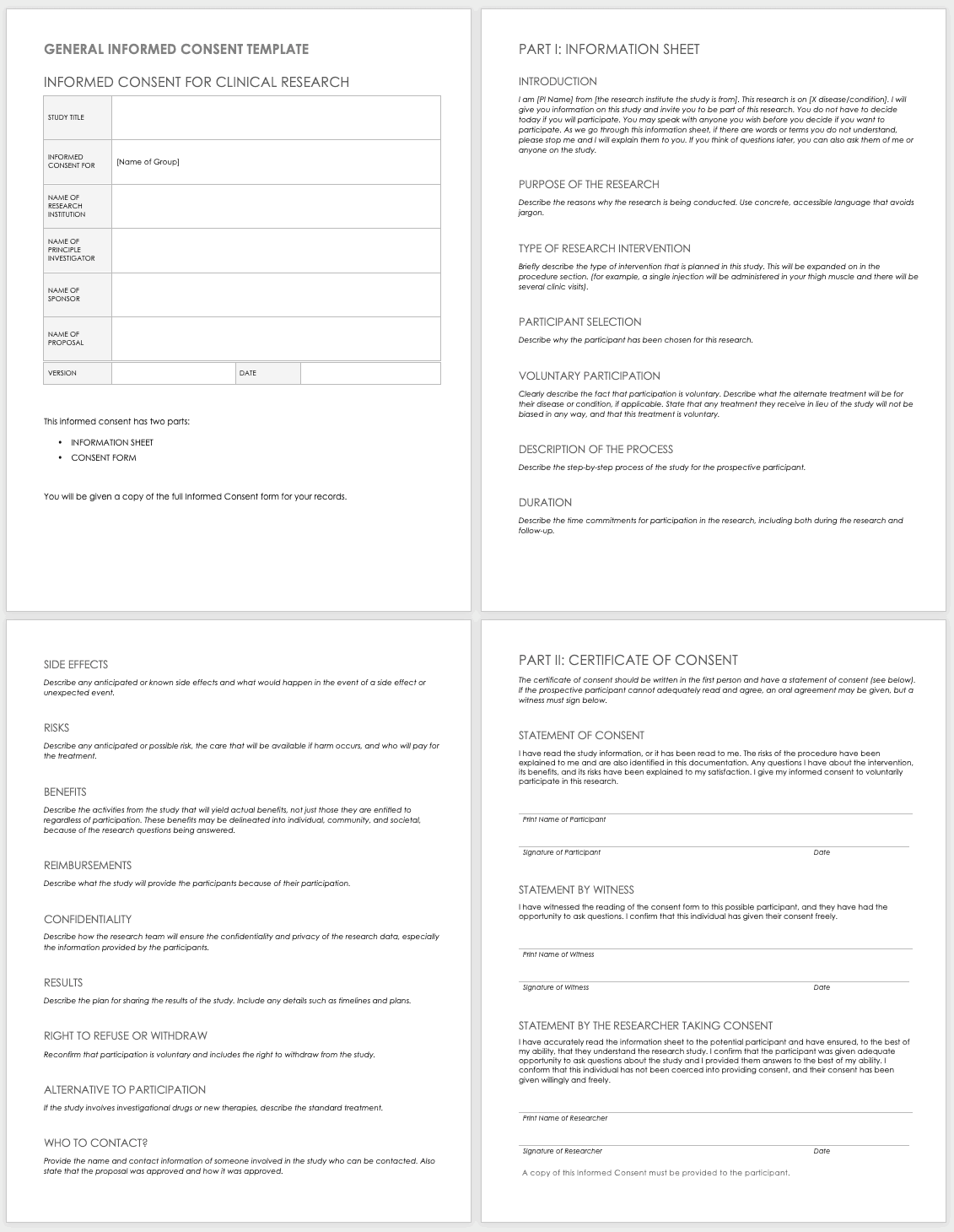
Download General Informed Consent Template - Word
Use the clinical trial template for medical research.

Download Informed Consent for Clinical Trials Template - Word
Eligibility Criteria (Inclusion/Exclusion) Checklist

Eligibility criteria are an essential part of clinical trials. They define the population under investigation.
Inclusion criteria are the standards that participants must meet to enroll in the study. For example, in a study on a new diabetes medication, you would likely want participants who have already been diagnosed with diabetes.
Exclusion criteria specify the characteristics that disqualify participants from taking part in the research. For example, in the diabetes study above, the proposed diabetes drug may target a specific age demographic. One exclusion criterion could be a participant whose age falls outside of the range.
Download Eligibility Checklist Inclusion-Exclusion Template
Concomitant Medication Log Template
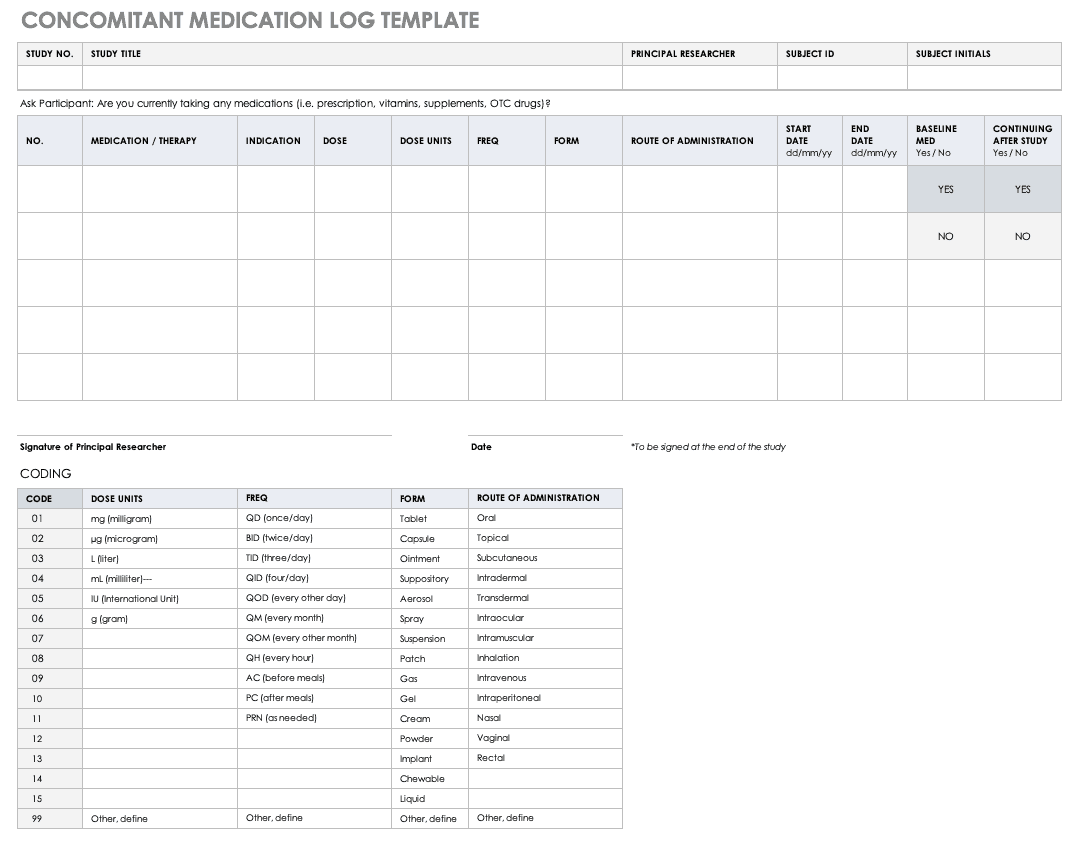
Properly documenting any medications that participants are taking is imperative to understanding the reactions occurring in their bodies, as well as what could spur adverse and severe adverse events during the study. Fill out a concomitant medication log for every participant and account for everything participants take, even seemingly innocuous items like multivitamins.
Download Concomitant Medication Log Template
Adverse Event Form
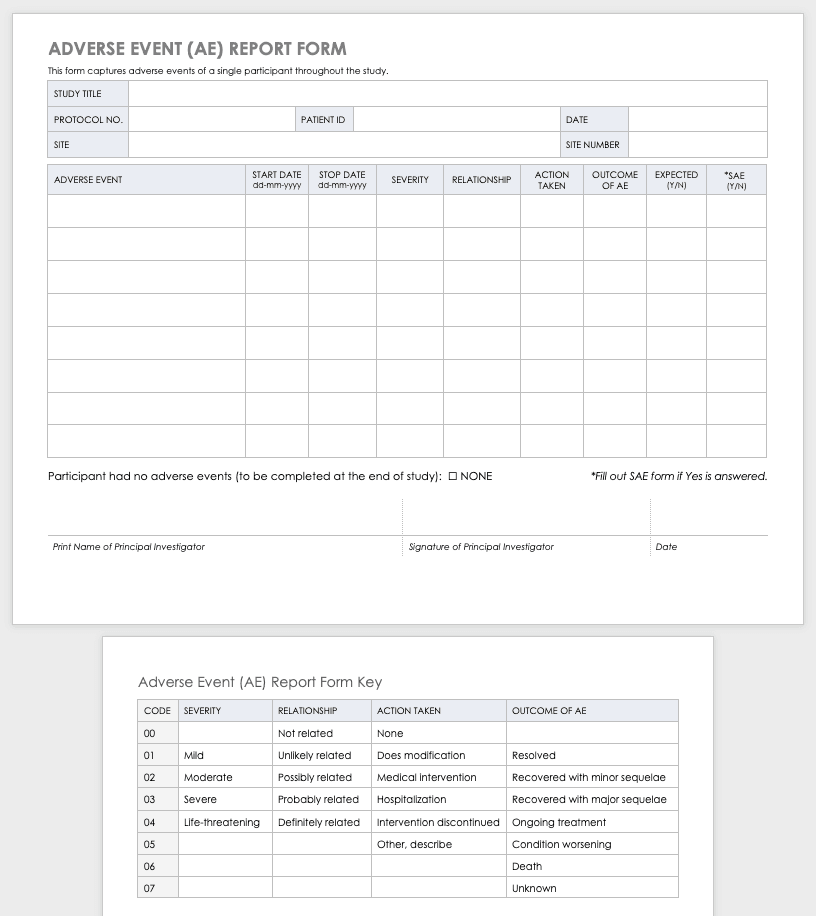
Clinical research can result in complications for the participants and trigger an adverse or severe adverse event. An adverse or severe adverse event is when participants in a clinical trial have negative medical symptoms that can be shown in laboratory or physical testing. Each participant in a clinical trial should have an adverse event log that tracks any adverse events through the duration of the study.
Download Adverse Event Form Template
Severe Adverse Event Form

A severe adverse event (SAE) is a special case of an adverse event in which the outcomes are acute. Examples of SAEs include death, life-threatening complications, or anything leading to immediate hospitalization, physical disability, or congenital abnormalities. Log SAEs in the AE form, but fill out an additional SAE form.
Download Severe Adverse Event Form Template
Word | PDF | Smartsheet
Post-Clinical Study Research Documentation and Templates
After you complete or terminate a clinical trial, you should prepare several additional documents. Here are some examples of this documentation:
- Investigational Product Accountability Log: You generally provide an accountability log to the authorities that tracks drug products to show product disposition and accountability per participant. It also helps you track the drug product stock and any imbalance at the end of the study.
- Investigational Product Destruction: Due to regulations governing the proper disposition of investigational products in clinical research, you must properly dispose of products left at the end of a study (as evidenced by the product accountability log). This form describes and ensures that you have properly handled any leftover products.
- Close-out Checklist/Report: A study close-out checklist and report helps ensure that you complete all closing procedures, archive the paperwork, and resolve electronic data.
Clinical Study Summary Report Template
Assemble the summary report at the end of a study to get results into the sponsor’s or public’s hands while you complete the full report. A summary report is typically about 2-3 page-long document that encompasses the highlights from the trial.
Download Study Summary Report Template - Word
Clinical Study Report (Full) Template

The full clinical study report (CSR) encompasses all aspects and details of the research you’ve conducted. It is not a sales or marketing tool; instead, it is a scientific report details the methodology and shows scientific rigor.
Download Clinical Study Report Template - Word
Public Links and Resources for Clinical Trials
The following are publicly available resources, tools, and links for clinical trial practitioners and principal investigators:
- PROMIS: Patient-Reported Outcomes Measurement Information System (PROMIS) software gives clinicians health status patient measures that are physical, mental, and social patient-reported metrics. Funded by the National Institutes of Health (NIH), PROMIS can be used in clinical trials as measures of conditions and disease and as a comparison to the general population. The measures in PROMIS are free to administer on paper, by computer (computer adaptive tests), or with an app. The computer adaptive tests may be conducted on REDCap, Assessment Center, or Epic.
- REDCap: REDCap (Research Electronic Data Capture) is an electronic data capture system that works on browsers to develop research databases. It was developed at Vanderbilt University to support clinical research data collection and is a free resource to nonprofit organizations. It is limited to organizations joining the REDCap consortium and is not open-source or available for commercial use.
- Good Clinical Practice (GCP) Training: GCP is an international quality standard designed for use by staff involved in clinical trials. The guidelines for this are from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). These regulate the ethical guidelines, documentation, record keeping, training, facilities, technology, and inspections. The purpose of these guidelines is to keep clinical trials scientifically rigorous and to delineate the roles and responsibilities of research staff. The National Institutes of Health administers training for GCP.
- Quality Management Study-wide Review Tool: Developed by the NIH, this review tool is for PIs and study teams to manage their quality reviews, and may be customized for unique studies.
- Quality Management Subject Review Tool: Also developed by the NIH, this review tool provides study teams the structure for review of participant data, and may be customized for the unique study. This should be developed in concert with the DSMP.
- AccrualNet: AccrualNet is sponsored by the National Cancer Institute (NCI), and offers advice and training to staff on how to recruit study participants.
- Regulatory Education for Industry (REdI): The FDA offers a Clinical Investigator Training Course for researchers conducting investigational new drug (IND) or device exemption (IDE) studies.
- ResearchMatch: Available to volunteers and researchers affiliated with the NIH Clinical and Translational Science Award (CTSA) program, this site helps match prospective participants with specific studies.
- Grant Policies and Guidance: The NIH and National Center for Complementary and Integrative Health (NCCIH) offer links to many resources that are policy- and grant-specific to the NIH and NCCIH, updated regularly.
- Protocol Amendments: The NIH and NCCIH offer regularly updated guidance for NIH policy and protocol changes.
- Clinical Terms of Award for Human Subjects Research: The NIH and NCCIH offer guidance for clinical trial grant awardees for compliance.
- NIH Single IRB (sIRB) Policy for Multisite Research: The NIH offers a FAQ page for multisite research that includes policy, contract and application information, responsibilities, exceptions, and costs.
- Dictionary of Cancer Terms: The National Cancer Institute (NCI) offers a dictionary of cancer terms for researchers and laypersons. You can add this dictionary to your website as a widget.
- Informed Consent FAQs: The U.S. Department of Health and Human Services (HHS) and the Office for Human Research Protections (OHRP) offer a FAQ page about informed consent for researchers and lay persons.\Informed Consent Language (ICL) Database: The National Comprehensive Cancer Network (NCCN) offers a database to help write informed consents. This database is specific to medical conditions and different risk language.
Improve Clinical Trial Research with Smartsheet for Healthcare
Empower your people to go above and beyond with a flexible platform designed to match the needs of your team — and adapt as those needs change.
The Smartsheet platform makes it easy to plan, capture, manage, and report on work from anywhere, helping your team be more effective and get more done. Report on key metrics and get real-time visibility into work as it happens with roll-up reports, dashboards, and automated workflows built to keep your team connected and informed.
When teams have clarity into the work getting done, there’s no telling how much more they can accomplish in the same amount of time. Try Smartsheet for free, today.